
Background
With a population of 63.9 million people and divided in 18 administrative regions, France has the second largest medical device market in Europe.
France’s national healthcare system is managed by the government and the Parliament, while 25 health agencies (Agences régionales de Santé) are responsible for the healthcare system at a regional level.
France provides a mandatory social health insurance system (Assurance Maladie Obligatoire – AMO) that covers the entire population. It’s financed through employers and employees’ payroll contributions and taxes. However, it does not cover every healthcare cost – that’s why 90% of the population has voluntary/complementary private health insurance, which covers the remaining expenses. For families with annual incomes below a fixed threshold, the government provides free complementary cover (Couverture Maladie Universelle Complémentaire - CMU-C).
Around 55% of hospitals are in the public sector (public and private not-for-profit). Public funding is applied to both private and public sectors, although tariffs differ.

Who are the decision makers/influencers?
National Level – Health Technology Assessment Organizations
- Agence Française de Securité Sanitaire des Produits de Santé (AFSSAPS, or French Health Products Safety Agency):
The French Health Products Safety Agency is responsible for all safety decisions concerning health products and for monitoring products once released in the market. Their 3 core missions are scientific evaluation, laboratory control and advertising control.
- Haute Autorité de Santé (HAS, or French National Authority for Health):
The French National Authority for Health aims to improve patient care and provide more consistency within the healthcare system. Their activities range from assessment of drugs, medical devices and different medical procedures to the publication of guidelines and accreditation of organizations.
This organization is an independent public body with financial autonomy – and report its activities to the Government.
- Commission Nationale d’Evaluation des Dispositifs Médicaux et des Technologies de Santé (CNEDiMTS, or National Commission for the Evaluation of Medical Devices and Health Technologies):
Part of the HAS, this organization is responsible for providing scientific opinions related to the use and interest of medical devices and non-drug healthcare products.
Reimbursement and pricing approval process
Approval
The first step to ensure that a medical device has market access is to gain certification by a notified body. In Europe, a CE mark ensures that medical devices meet the European Directives requirements.
Reimbursement
First Step - DRG Funding
The DRG Funding is a scheme of reimbursement applied to both public and private sectors. Most devices are included in this tariff and are funded by the National Health Insurance.
The role of COMEDIMS, a subcommittee set in every hospital/group of hospitals, is to enlist and decide which devices to buy for their respective hospitals. The devices not included in this funding are usually considered as innovative and/or costly devices. For these last two categories a separate budget is allocated, for which funding is usually temporary, until the device is included in the DRG tariff.
Second Step - Enlistment on the LPPR, a positive reimbursement list
The reimbursement by the MHI (Mandatory Health Insurance) for a device in ambulatory care or for a device that is too expensive for the DRG tariff is enlisted on a ‘positive list’ (LPRR - Liste des Produits et Prestations Remboursables).
There are 2 pathways to be enlisted:
- Listing under “generic line”
Generic lines represent a class of products, their use, and technical characteristics without mentioning any company or brand name. If a medical device conforms with the LPRR generic line description, it doesn’t need to go through a CNEDiMTS evaluation. The manufacturer only needs to label the products with a LPRR code and declare it to the Health Products Safety Agency for future monitoring. This means the device will be reimbursed by the MHI at the existing tariff.
- Listing under the medical device own trade name
In this list, products are designated with their own brand name. It usually happens when the device is innovative/with different specification and are presumed to impact health care expenditure. This listing is also recommended when the device needs a specific follow-up due to associated safety issues.
CNEDiMTS requires the manufacturer to submit 2 dossiers: a technical dossier and an economic dossier. The technical dossier includes technical description of the technology and its use, the severity of the condition, clinical benefits and alternatives. The economic dossier includes the price or tariff required for reimbursement, sale forecasts and price justification, as well as a breakdown of costs, from manufacturing to distribution. A cost/effectiveness analysis may be submitted, in both dossiers.
Afterwards, a two-step process is implemented:
1. Technical assessment by CNEDiMTS will verify:- If a trade name listing is appropriate according to their characteristics;
- If the service provided by the device is sufficient to guarantee reimbursement;
- What is the added value for the patient
- Number of patients who might get benefits from the device
- The Economic Committee for Health Care Products (CEPS: Comité Economique des Produits de Santé), in negotiation with the manufacturer, sets a tariff for reimbursement by the MHI.
If there are many trademark devices with similarities, the CEPS can consider creating a new generic description to set a common tariff. Eventually reimbursement is submitted to set up registry and produce further clinical evidence.
According to law, the time for the process should be 180 days but could also take longer.
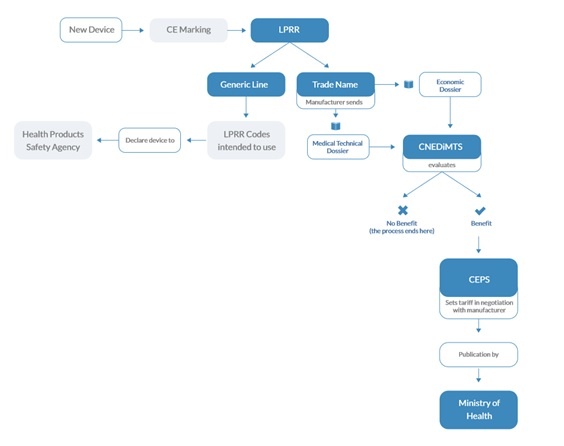
Figure 1, Approval & Reimbursement Process when enlisted on the LPRR
The future
If the medical device is only used within a medical procedure that is not coded for reimbursement, the new procedure must be enlisted to obtain reimbursement in an ambulatory or hospital setting.
If the medical procedure doesn’t need to be coded for reimbursement, the French National Authority for Health (HAS) only needs to submit an assessment report and conclusions on the safety and efficacy of the new device. These are transmitted to the CEPS and Ministry of Health.
Based on the approval, the Committee of Grading of Medical Procedures (CHAP: Commission De Hiérarchisation des Actes Professionnels) fixes reimbursement tariffs after negotiating with unions of health professionals.
